1 HCAM - Hidropathy Clustering Assisted Method
1 HCAM - Hidropathy Clustering Assisted Method
Prediction of the native structure of newly characterised proteins is a central problem
in molecular biology. This is why development of theoretical methods for identifying
folding of polypeptide chains has become an important field of research over the past few
years, permitting prediction algorithms based on many new approaches to be developed. Water
soluble globular proteins are known to fold into structures in which hydrophobic residues
are disposed on the inside while lateral chains of hydrophilic residues are exposed to the
solvent. In native conformation, apolar residues tend to be disposed in the core, while
polar residues tend to occupy the surface of the protein. Moreover, these proteins typically
have an abundance of secondary structures such as α-helices and β-sheets connected by
flexible loops that often play a role in determining biological diversity of function and
activity. Despite the many interactions occurring between distant parts of the amino acid
chain, a large variety of different protein sequences seem to fold into similar structural
motifs and each structural element has distinct fields of preference for different amino
acids, suggesting the presence of a finite number of folds, even thought the astronomical
variety of possible combinations of residues composing polypeptide chains. In the course of
evolution, the three-dimensional structure of certain folds seems conserved to a greater
extent than the corresponding amino acid sequence, since substitution of certain residues
that tend to stabilise the folding motifs may be compensated by other substitutions that
give the structure stability. Moreover, several recurrent structural motifs retain specific
biological functions. Since our capacity to predict the three-dimensional structure of a
protein is limited by lack of a general paradigm, most prediction methods have focused on
identification of both secondary and tertiary structure of proteins by computational
approaches. Although several computational methods made a great effort to predict the whole
proteins structure at high atomic resolution, template based search by sequence similarity
(comparative modelling) and conformational search (ab initio modelling) are the strategies
so far mostly used to obtain the tertiary structure of amino acid sequences and are mainly
directed at predicting certain conserved structural sub-domains, as well as fragment-search
based methods that focused on the prediction of well known super-secondary folds. Several
properties shared by certain protein structural motifs could suggest strategies for the
prediction of large populations of recurrent folds. For example, a case study based on
exhaustive analysis of crystallized protein structures indicates that the main building
blocks of globular proteins actually consist of closed loops of standard length. Protein
structure can therefore be seen as a compact array of closed loops that can be grouped into
two main sub-populations with 11-15 and 25-35 amino acids, respectively, that follow each
other in strict linear order (see Figure 1). The presence of certain sequence motifs typical
of closed loops enabled certain “prototypes” common to proteins of many bacteria to be
identified, suggesting the existence of a proteomic code. The extremities of closed loops
are often in close contact with each other (distance C-C < 10 Å) and are characterised by
3-5 hydrophobic and/or non polar residues, often corresponding to “hydrophobic folding units”
(HFUs) of globular proteins.
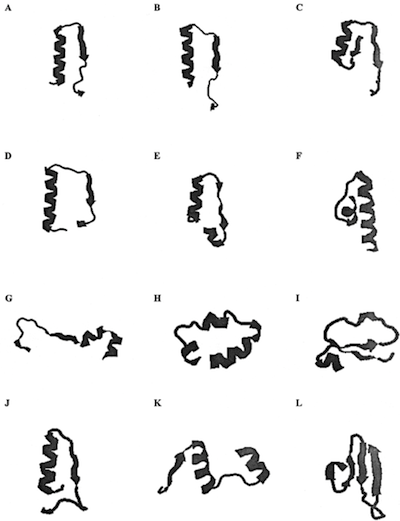
Figure 1. Chain trajectories of the matching segments extracted from
the PDB_SELECT database (Hobohm and Sander, 1994). A, protein 1b0u, chain A (match 26);
B, 1f2t, chain B (match 15); C, 1cs1, chain A (match 12); D, 1qhf, chain A (match 11);
E, 1qap, chain A (match 10); F, 1di1, chain A (match 10); G, 1ds1, chain A (match 10);
H, 1nf1, chain A (match 9); I, 8ohm, chain A (match 9); J, 1d4c, chain C (match 9);
K, 6gsv, chain A (match 9); L, 1guq, chain A (match 9).
Figure fetched from:
Igor N.Berezovsky, Valery M.Kirzhner, Alla Kirzhner, Vladimir R.Rosenfeld and Edward N.Trifonov.
Closed loops: persistence of the protein chain returns.
Protein Engineering vol.15 no.12 pp.955–957, 2002.
Typical closed loops of 25-35 amino acids exhibit similar features for small and large proteins. The
selective pressure common to closed loops is not only reflected in structure but also in the
typical distance between hydrophobic residues, which is equal to the distance between the
two extremities of the folding motif. Prediction of these extremities can therefore be
obtained by means of physicochemical parameters such as the Kyte-Doolittle hydropathy scale,
which is particularly indicated for revealing certain properties of globular proteins.
Hydropathy scales are currently used to inspecting the hydrophobic character of proteins in
order to reveal families of transmembrane helices, potential antigenic sites and regions
that could be exposed on the protein surface. Moreover, the prediction power of hydropathy
profile lies in the possibility of clarifying evolutionary relationships more distant than
those obtained by comparison of amino acid sequences, but it has not yet been narrowed to
the detection of secondary structure profiles shared by recurrent super-secondary structure
motifs of both globular and transmembrane proteins. Here we endeavour to combine several
analyses for obtaining fingerprints for identification of position and length of secondary
structures placed in protein elementary building blocks, including closed loops and other
typical sub-domains of polypeptide chains. Thus, we introduced the notion of Hydrophobic
Assistance For Secondary Structure Detection and we decided to define our algorithm as
“Hydropathy Clustering Assisted Method” (HCAM) because of the main contribution given by
hydropathy analysis. HCAM results can be read as a whole protein prediction map, but the
association of secondary structures with previously collected hydropathy patterns should
lead toward targeted super-secondary motifs identification. In secondary structure prediction,
any ambiguities caused by overlap of hydropathy patterns of certain types can be resolved
by complementary statistical analysis based on confidence levels derived from the frequencies
of particular amino acid motifs and by use of hydrophilic and hydrophobic residue patterns
(binary patterns) that play an important role in protein architecture and can be divided in
two classes in relation to whether they preferentially form α-helices or β-sheets.
The sequences of each class of protein building motifs were processed by multiple alignment
analysis using the Gonnet substitution matrix in the programme ClustalW (see Figures 2a and
2b). Secondarily, the secondary structure members (α-helices, β-strands and coils) of each
class were partitioned in clusters containing amino acid sequence motifs and the corresponding
hydropathy values. Loop sequences linking motifs such as βαβ barrels, α-hairpins, αβ-barrels,
etc. were partitioned.
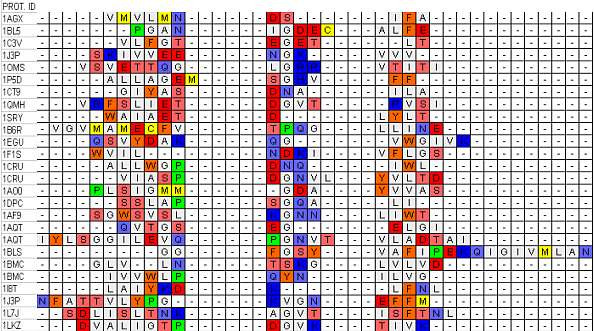
Figure 2a. Example of multiple alignment analysis by Gonnet
substitution matrix of β barrels collected in our database. Each group of residues columns
corresponds with good approximation to a separate type of secondary structure.
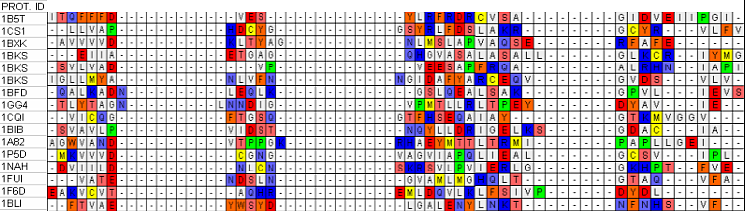
Figure 2b.Example of multiple alignment analysis by Gonnet
substitution matrix of βαβ barrels collected in our database. Each group of residues columns
corresponds with good approximation to a separate type of secondary structure.
1.2 Hydropathy profile analysis
The Kyte-Doolittle scale combined with a 5-amino acid sliding window is a useful instrument for
revealing the topological disposition of secondary structures inside several building blocks.
For example, careful observation of the hydropathy profile of prokaryotic globular proteins
showed many β-barrels with two lateral hydrophobic “icebergs” (positive hydrophobic domains)
3-6 amino acids long, flanking a central negative area of variable length, generally not less
than 3 amino acids; the two icebergs often correspond roughly to residues forming β-strands of
β-barrels, whereas the negative central domain is often associated with hairpin folds. Visual
analysis of βαβ barrels also revealed hydrophobic icebergs associated with β-strands and a
ragged central domain, associated with α-helices, having positive peaks separated by minima,
sometimes less than zero (see Figure 3a-b). Small continuous hydrophobic domains may also
separate icebergs from the ragged central domain; these domains often correspond to loops
connecting β-strands and α-helices. Similarly, observation of the profile of α-hairpin motifs
often revealed two ragged domains of peaks (α-helices) surrounding a domain with negative
hydropathy (hairpin). However, certain domains corresponding to α-helices may sometimes be
mistaken for hydrophobic icebergs erroneously associated with β-strands, due to a contiguous
series of hydrophobic residues, especially Val, Ala and Leu, within the structural motif
(see Figures 4a-d). Hydrophobic portions corresponding to coils can also sometimes have positive
values (presence of Gly and Ala), but hydropathic peak form is generally different from that of
icebergs of β-strands. Thus, profile pattern is not unambiguous, but nevertheless provides more
or less detailed information on the position of certain structural motifs.
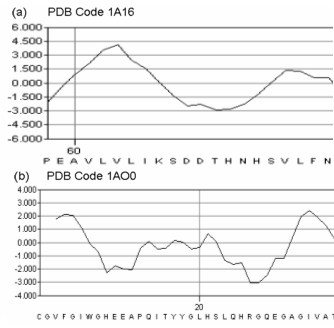
Figure 3a-b.Hydropahty profile of both a typical β-barrel (a)
and a typical βαβ barrel of our database.
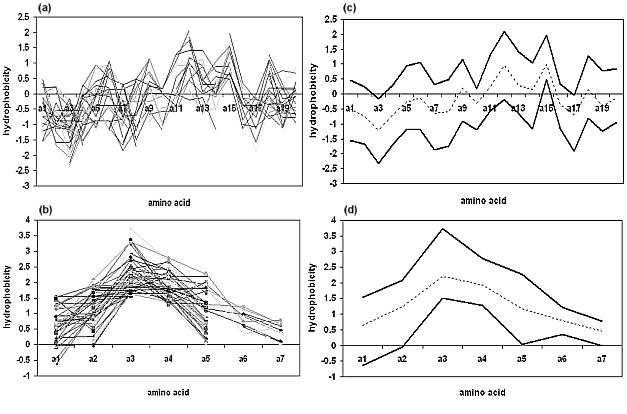
Figure 4a-d.Left side: an example of clusters of hydrophobic
profiles calculated from amino acidic sequences corresponding to (a) α-helices and (b) β-strands.
Right side: example of secondary structure identification patterns obtained from cluster analysis of
(c) α-helices and (d) β-strands hydrophobicity.
1.3 Statistical analysis of amino acid sequences
To compensate for the ambiguities encountered in hydropathy profiles, a complementary analysis
of secondary structures, based on amino acid frequencies in sequence clusters, was conducted.
We chose to calculate the frequency of 20 x 20 combinations of dipeptides and the frequency of
a large amount of tripeptides observed in structural clusters formed by α-helices, β-strands,
coils and β-hairpins. Permutations of a sequence of n amino acids were considered.
If aa1, aa2, aa3, ...,aai,..., aan are the
amino acids of the sequence, the linear permutations of the n-1 dipeptides will be: aa1
aa2, aa2 aa3, ..., aai-1 aai, ...,
aan-1 aan, and those of the n-2 tripeptides will be: aa1
aa2 aa3, ..., aai-1 aai aai+1, ...,
aan-2 aan-1 aan, where n ≥ 3.
1.4 Statistical analysis of binary patterns
Protein sequences can be represented as binary patterns of polar (p) and non polar (n) amino
acids. The linear sequence of amino acids can therefore have particular dispositions of polar
and non polar residues, which may promote formation of particular secondary structures. These
patterns have been used as a binary code for protein design, where the precise identity of
residue composing the binary sequence can vary with certain flexibility. We therefore used
binary patterns to define guidelines for identifying secondary structures in amino acid
sequences, calculating the frequency of the 23 combinations of binary motifs obtained from the
linear permutations of tripeptides. The residues included in the category of polar amino acids
were: Arg, Lys, Asp, Glu, Asn, Gln and His, whereas non polar amino acids were: Phe, Leu, Ile,
Met, Val, Trp and Cys.
Analysis on an independent set of 150 globular proteins showed that the prediction method was
particularly sensitive for each class of secondary structure and demonstrated that HCAM can
also be used as a stand alone prediction program. Indeed, parameter Q3 (ratio of number of
residues correctly predicted to total number of residues identified by the method) for
elementary building blocks and closed loops was estimated at about 76% ± 5, while Q3 for the
entire protein sequences was estimate at about 73%± 5. Moreover, the predictive estimate of
secondary structure of building blocks motifs can considerably improve with manual intervention
by expert operators, based on multiple alignment consensus methods and visual analysis of peaks
of the overall hydropathy profile of the sequences: in this case Q3 may be as high as 81% since
the help provided by the method makes manual identification of hydrophobic domains simpler.
Although hydrophobic domains cause interpretative incongruence if viewed in the global
topological context of the polypeptide sequence, they may suggest the exact correspondence with
a secondary structural motif.
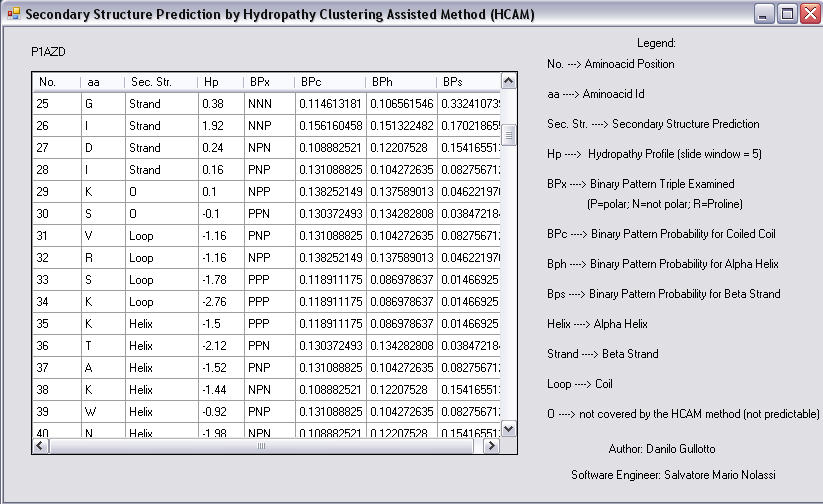
HCAM snapshots: